

Stingele Lab - Research
- Research
DNA-protein crosslink repair
DNA in every cell is damaged by tens of thousands of lesions every day. The challenge to maintain genome stability is further complicated by DNA lesions being very diverse in nature. Therefore, cells employ highly-specialized pathways, to detect, signal and repair different types of lesions.
The fundamental importance of genome maintenance is exemplified by germline mutations in DNA repair genes causing a range of human syndromes characterized by cancer predisposition, premature ageing, immunodeficiency and neurodegeneration. Moreover, somatic mutations of crucial DNA repair genes are responsible for the severe genome instability observed in cancer cells.
Importantly, this renders tumour cells sensitive to DNA damaging agents and to the inhibition of other repair factors, which can be exploited for targeted anti-cancer therapies. Nearly half of the current cancer treatment regimens involve treatment with agents inducing covalent DNA-protein crosslinks (including topotecan, doxorubicin, etoposide, carboplatin, oxaliplatin). DNA-protein crosslinks (DPCs) are highly toxic, because they are exceptionally large adducts, which block chromatin processes, such as transcription and replication.
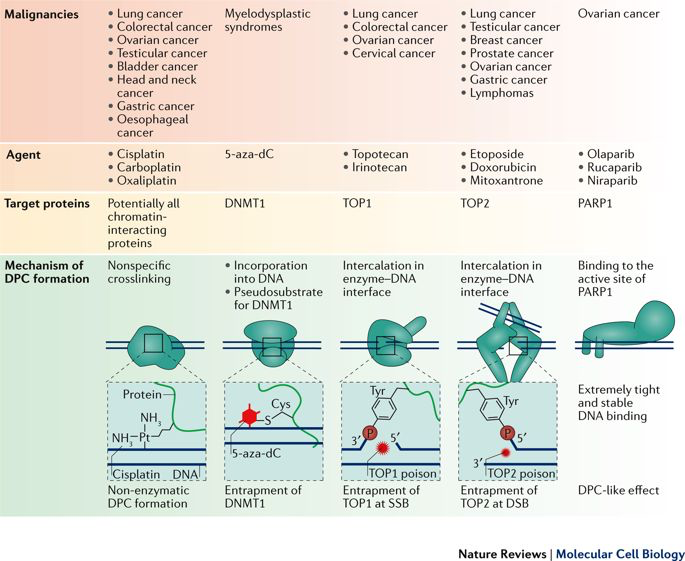
Reprinted by permission from Macmillan Publishers Ltd: Nat. Rev. Mol. Cell Biol. (Stingele et al., 18(9):563-573, 2017).
Until recently, it was assumed that these lesions were targeted by canonical DNA repair pathways such as homologous recombination (HR) and nucleotide excision repair (NER). This has changed with our discovery of a protease-based DPC-specific repair mechanism, which is conserved from yeast to humans and is essential for genome stability and tumour suppression. Our work established that cells possess specialized proteases (termed DPC proteases: SPRTN in mammals, Wss1 in yeast) that degrade the protein component of DPCs, which enables replication of DPC-containing DNA.