

Canzar Lab - Research
- Research
- CIDANE
- Ladder-seq
Finding the Invisible, and Improving Accuracy, in Genetic Transcription
High-throughput sequencing techniques create an avalanche of fragmented genetic data. Our lab uses advanced algorithms to reassemble that information and detect hidden patterns.
Higher organisms store their genetic material in the nuclei of cells as deoxyribonucleic acid (DNA). In a process called transcription, individual segments, the genes, are converted into messenger ribonucleic acids (mRNAs). Subsequently, the translation process produces proteins as the most important functional units.
Accurately reconstructing the basic protein patterns in the genetic blueprints of life can help us understand cellular biology and identify specific patterns in genes that produce diseases. The preferred method of converting samples of cellular RNA and other cellular featuers into genetic data is (RNA) sequencing, which creates hundreds of millions of small data fragments, known as reads. In our lab, we develop algorithms and open-source software that piece together genomic reads to molecular measurments and to more complex cellular patterns, to gain new insights into fundamental problems in biology and human disease.
For the development of accurate and efficient computational methods we combine techniques from combinatorial optimization and machine learning. For instance, we have developed exact and approximate algorithms for a graph coloring problem to increase the resolution of experimental protein structure data, used neural networks to project high-dimensional cellular measurments to an interpretable low-dimensional space, and extended dynamic time warping to the comparison of complex trajectories of, e.g., differentiating immune cells.
The software tools we develop address important biological and medical questions. In a collaboration with the Song lab (University of Pennsylvania) we have contributed to the discovery of the embryonic origin of adult neural progenitors, and the epitranscriptomic temporal control of mouse brain development; we have helped Tobias Feuchtinger at the pediatric university hospital of the LMU Munich to link TIM-3 expression to increased relapse risk in pediatric patients with acute lymphoblastic leukemia.
Computational methods for transcript reconstruction
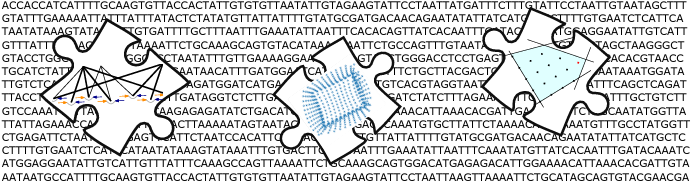
Piecing together the mass of highly fragmented data produced by RNA-seq into the full-length molecular sequences, known as transcripts, is similar to solving a jigsaw puzzle and is very difficult.
The technique pioneered by our group together with colleagues at Freie Universität Berlin (Germany) and Centrum Wiskunde & Informatica (The Netherlands), CIDANE, mixes techniques from machine learning and combinatorial optimization to reconstruct transcripts more accurately and comprehensively than previous methods.
“It is a big jigsaw puzzle. You want to figure out the big picture—these RNAs—but you only have these small fragments, these puzzle pieces.”
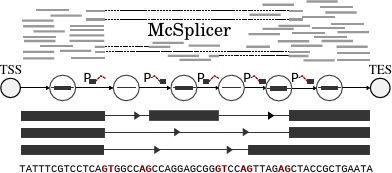
Instead of trying to computationally reconstruct individual outcomes of genetic transcription, our team, in collaboration with Heejung Shim at University of Melbourne, has developed a simplified probabilistic model of the underlying process itself. The model is based on the usage of basic informational blocks that make up transcripts. McSplicer uses an expectation-maximizaton algorithm to more accurately and efficiently estimate these usages. The model is able to describe multiple effects of point mutations using few, easy to interpret parameters, as we illustrate in an experiment on RNA-seq data from autism spectrum disorder patient.
 Original Publication: Bioinformatics, 2021.
Original Publication: Bioinformatics, 2021.
The problem with established high-throughput RNA sequencing technologies is that information was often lost along the way. Our team has now presented Ladder-seq, in which we combine changes to the sequencing protocol with tailored algorithms to recover some of this information. Figuratively speaking, an extra step in the experiment adds color information to the genetic puzzle pieces that our tailored algorithms use to piece together individual parts of the puzzle with greater precision than previously possible. With Ladder-seq we were able to decode the function of regulatory units of neural stem cells in the brains of mice.

Original Publication: Nature Biotechnology, 2022.
Efficient algorithms for single-cell omics
Experimental methods for sequencing DNA or RNA of single cells have transformed biological and medical research. To exploit the full potential of single-cell genomics technologies, we devise and engineer algorithms that can narrow the gap between the scalability of current analytical methods and the sheer volume of the data being produced. At the same time, the computational methods that we develop link multiple modalities measured by emerging technologies, including genome, transcriptome, epigenome, and proteome, in a biologically meaningful manner, taking into account the spatial context in tissues.
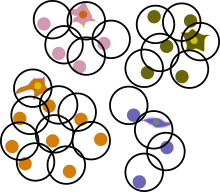
In practice, scRNA-seq analysis methods are often run on a smaller subset of the data whose enormous size exceeds the capabilities of existing analysis methods. We have developed method Sphetcher that efficiently picks representative cells that accurately capture the geometry of the transcriptional space occupied by the original data. The resulting sketch of single cells highlights rare cell types, facilitates visualization and sharing of large datasets and accelerates downstream analyses such as trajectory inference.
Original publication: iScience, 2020.
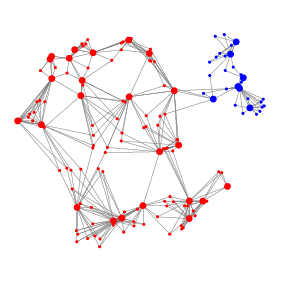
Another fundamental task in scRNA-seq analysis is the identification of transcriptionally distinct groups of cells. We have proposed method Specter whose core algorithmic innovation allows it to cluster a dataset comprising 2 million cells in under half an hour. The ensemble learning approach implemented in Specter is able to utilize multimodal omics measurements such as RNA and surface protein expression to resolve subtle transcriptomic differences between subpopulations of, e.g., memory T cells.
Original publication: Genome Research, 2021.
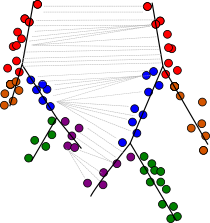
scRNA-seq can illuminate the dynamic changes in gene expression underlying biological processes such as differentiation and development. We have developed Trajan, a novel method to compare complex trajectories from two conditions. Trajan aligns trajectories by solving a variant of a constraint matching problem for which we have established a theory that links it to dynamic type warping, a fundamental tool for the comparison of temporal sequences in, e.g., speech recognition. In an alignment of single-cell trajectories describing human muscle differentiation and myogenic reprogramming, Trajan identifies the correspondence between core paths without prior information, from which we are able to reproduce recently reported barriers to reprogramming.
Original publication: RECOMB, 2019.
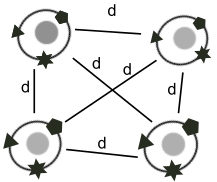
t-SNE and UMAP are the two most widely used embeddings used to visualize multimodal omics data. Typically, these methods are applied to each modality independently, and the resulting embeddings then need to be reconciled manually. We have extended both algorithms to compute an embedding of cells that takes into account multiple modalities simultaneously, which greatly facilitates the visual interpretation of multimodal omics data. Our two algorithms j-SNE and j-UMAP are freely available in software package JVis.
Original publication: Genome Biology, 2021.