

Wolf Lab - Research
- Research
- Monogenic Kidney Diseases
- Reproductive Biology
- Translational Animal Models
- Xenotransplantation
Molecular Basis and Consequences of Monogenic Kidney Diseases
Inherited kidney diseases represent the fifth most common cause of end-stage renal disease after diabetes, hypertension, glomerulonephritis, and pyelonephritis. Nearly all children and at least 10% of adults who receive renal-replacement therapy (RRT) have an inherited renal disease. In most cases, inherited kidney diseases are severe diseases with insufficient knowledge and insufficient treatment options available.
Our research activity is currently focused on thrombotic microangiopathy (TMA), which is a severe glomerulopathy, and on autosomal-dominant tubulointerstitial kidney disease – UMOD (ADTKD-UMOD).
TMA is a morphological symptom occurring in numerous glomerulopathies like in hemolytic uremic syndrome (HUS), as side effects of drugs or in preeclampsia. Common pathogenic mechanism of TMA irrespective of the cause is a primary endothelial injury. In the Munich ENU mutagenesis project, a mutant mouse line with glomerular specific TMA was generated and is currently investigated to evaluate the pathophysiology of dysfunctional endothelium in TMA.
ADTKD-UMOD is a tubulopathy caused by amino-acid changing mutations of the UMOD gene leading to maturation and secretion defects of the glycoprotein uromodulin (UMOD). It is a slowly progressive renal disease leading to end-stage kidney disease between an age of 20 and 70 years. The prevalence in humans is estimated 1.7 per million population, and 0.7 per 1,000 RRT patients. Using our two ENU-derived mutant mouse models for ADTKD-UMOD, the lines UmodA227T and UmodC93F, we showed that gain-of-toxic-function UMOD mutations lead to pronounced protein maturation retardation in the enlarged endoplasmic reticulum (ER) and decreased secretion of mutant and wild-type UMOD protein. As a consequence, dysfunction of the thick ascending limb of Henle’s loop (TAL) – the tubule segment where UMOD is selectively expressed – and later on, renal dysfunction occurs, leading to pronounced clinical symptoms. We recently identified, that UMOD maturation retardation induces chronic ER stress and induction of unfolded protein response in ER, and causes secondary consequences in other organelles in TAL, like in mitochondria and peroxisomes. In detail, mitochondrial UPR and mitophagy occurs, mitochondrial fission is reduced, and mitochondrial biogenesis is disturbed, mostly due to disturbed maturation and processing of NRF1, a transcription factor regulating pathways in the citric acid cycle and the mitochondrial respiratory chain. The activation of the LKB1-AMPκΒ pathway indicates alterations in TAL energy metabolism in ADTKD-UMOD. The NF-κB signaling pathway is activated. Future studies will search novel targets for therapeutic strategies that are so far not available for ADTKD-UMOD.
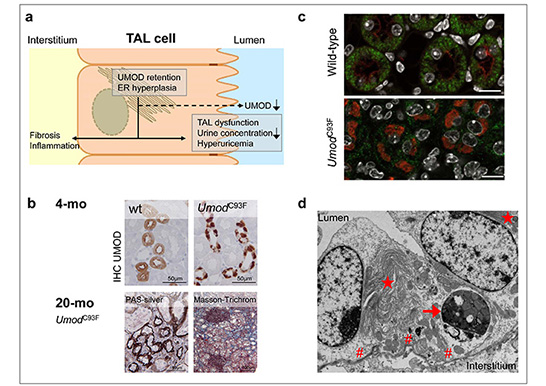
a) Scheme of the ADTKD-UMOD pathogenesis (modified according Rampoldi et al., 2011). Amino acid-changing mutation of the UMOD gene cause UMOD maturation retardation with UMOD retention in the hyperplastic endoplasmic reticulum (ER), associated with reduced UMOD excretion in urine, TAL dysfunction, reduced urine concentration ability of the kidney, hyperuricemia and morphological kidney alterations with interstitial inflammation and fibrosis. b) Morphological features of the ADTKD-UMOD mouse model: Young-adult UmodC93F mutant mice in an early ADTKD-UMOD disease status exhibiting paranuclear UMOD accumulation in TAL cells. 20-month-old UmodC93F mutant mouse in progressed ADTKD-UMOD disease status having renal tubular atrophy, interstitial fibrosis and lymphocytic infiltrates. c) Immunofluorescence analysis of SDHA (green, mitochondrial marker) and